Strongly recommended by AAD and AAAAI guidelines as a systemic therapy for moderate-to-severe atopic dermatitis in adult patients1,2
ADULT
Efficacy Results
Visible skin clearance and itch relief with Adbry®3-5
On this page:
Real-World Evidence
Systematic review and meta-analysis*

*Systematic review of observational studies (until July 2024) from PubMed and EMBASE databases screened 558 studies, of which 19 were included in the review.
A meta-analysis conducted statistical pooling of relevant data.
Limitations: Limitations from real-world data should be considered, including the absence of an unbiased control group and that the frequency of dosing could not be addressed. There are differences in reporting of efficacy and safety outcomes and the time for evaluation, and AE surveillance and reporting in the real world are not as rigorous as RCTs.

Limitations: This is a real-world study and limitations include the limited number of patients with ~6-month registry visits. Only persistent patients are included in the current analysis.
*Interim analysis of US adults (n=259 at baseline; n=81 with follow-up visit between 5-9 months from Adbry initiation) in the prospective, non-interventional CorEvitas® registry who initiated Adbry from February 2022 to May 2023.
Pivotal Studies
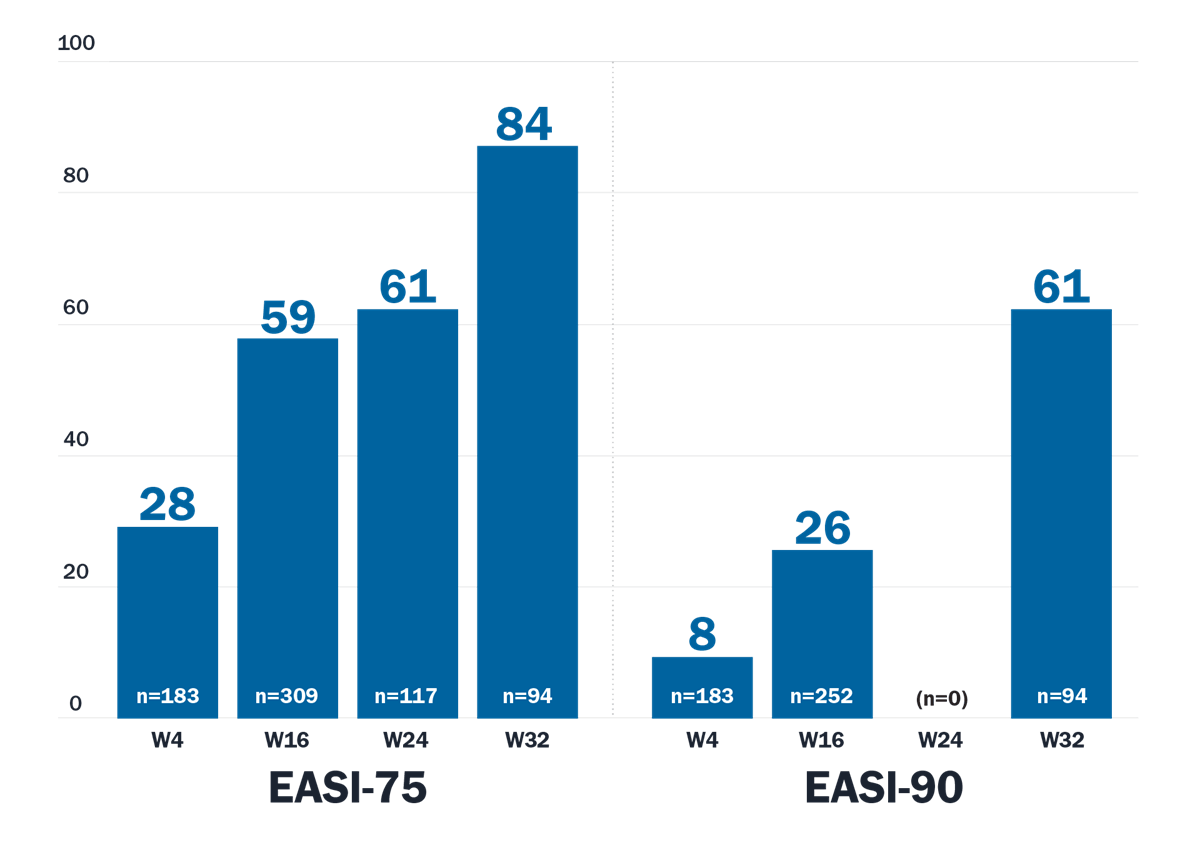
Adbry demonstrated significant improvements in skin clearance and lesion extent and severity3,8

*Patients who received rescue treatment or with missing data were considered nonresponders.
†Percentage of patients who achieved response.
‡Patients with baseline worst daily pruritus NRS (weekly average) score ≥4.
Trial designs: The efficacy and safety of Adbry was assessed in 3 randomized, double-blind, placebo-controlled trials with a total of 1934 patients 18 years of age and older, with moderate-to-severe atopic dermatitis not adequately controlled by topical medications. In all ECZTRA 1, 2, and 3 trials, subjects received subcutaneous injections of Adbry 600 mg or placebo on Day 0, followed by Adbry 300 mg or placebo every other week for 16 weeks. In ECZTRA 3, subjects also used TCS as needed. All ECZTRA 1, 2, and 3 trials assessed the primary endpoints of the proportion of subjects with an IGA 0 or 1 at Week 16 and the proportion of subjects with EASI-75 at Week 16. Secondary endpoints included the reduction of Worst Daily Pruritus NRS (weekly average) of at least 4 points on the 11-point itch NRS from baseline to Week 16.

Patients had significant disease severity at baseline.
*Proportion of patients.
EASI=Eczema Area and Severity Index; IGA=Investigator’s Global Assessment; NRS=Numeric Rating Scale; q2w=every 2 weeks; q4w=every 4 weeks; TCS=topical corticosteroid.
Maintenance of Response
With Adbry, your patients can achieve lasting disease control for your patients at Week 32 and Week 528
Patients receiving Adbry + TCS (as needed) who achieved EASI-90 response3*†
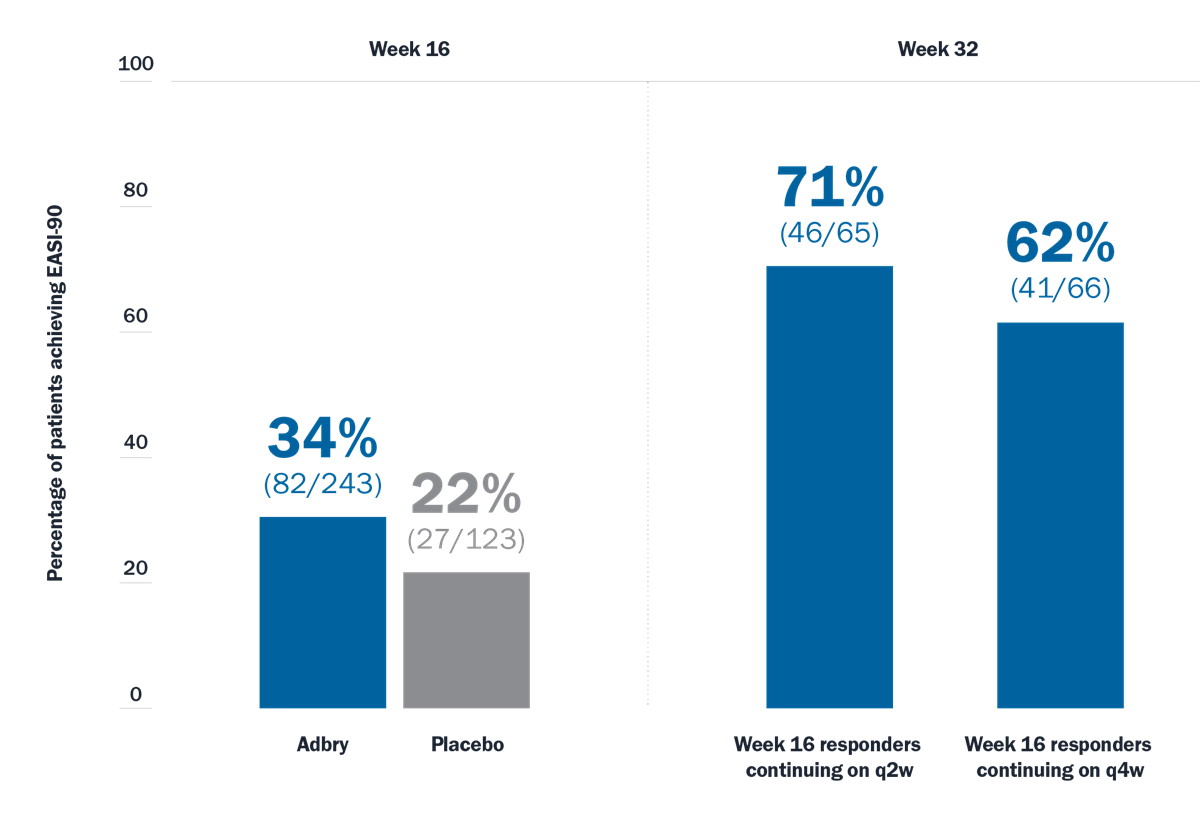
Limitations: EASI-90 was a prespecified endpoint at Week 16 and Week 32. Analyses were not adjusted for multiplicity and conclusions should be made with caution.
*Responders were defined as subjects with an IGA 0 or 1 (“clear” or “almost clear”) or EASI-75 at Week 16. At Week 16, responders were re-randomized to Adbry 300 mg q2w + TCS or Adbry q4w + TCS for another 16 weeks.
†Subjects who received rescue treatment or with missing data were considered nonresponders.
q2w=every 2 weeks; q4w=every 4 weeks; TCS=topical corticosteroid.

Itch Response
Itch reduction was observed with Adbry at 48 hours, 16 weeks, and up to 3 years3,4,8
Worst daily pruritus NRS (≥4-point reduction) at Week 16
ECZTRA 1 (P=0.002): Adbry 300 mg q2w: 20% (n=594); Placebo: 10% (n=194)
ECZTRA 2 (P<0.001): Adbry 300 mg q2w: 25% (n=563); Placebo: 9% (n=192)
ECZTRA 3 (P=0.039): Adbry 300 mg q2w + TCS: 46% (n=240); Placebo + TCS: 35% (n=123)
ECZTRA 1 + 2† MONOTHERAPY POOLED DATA (q2w)3
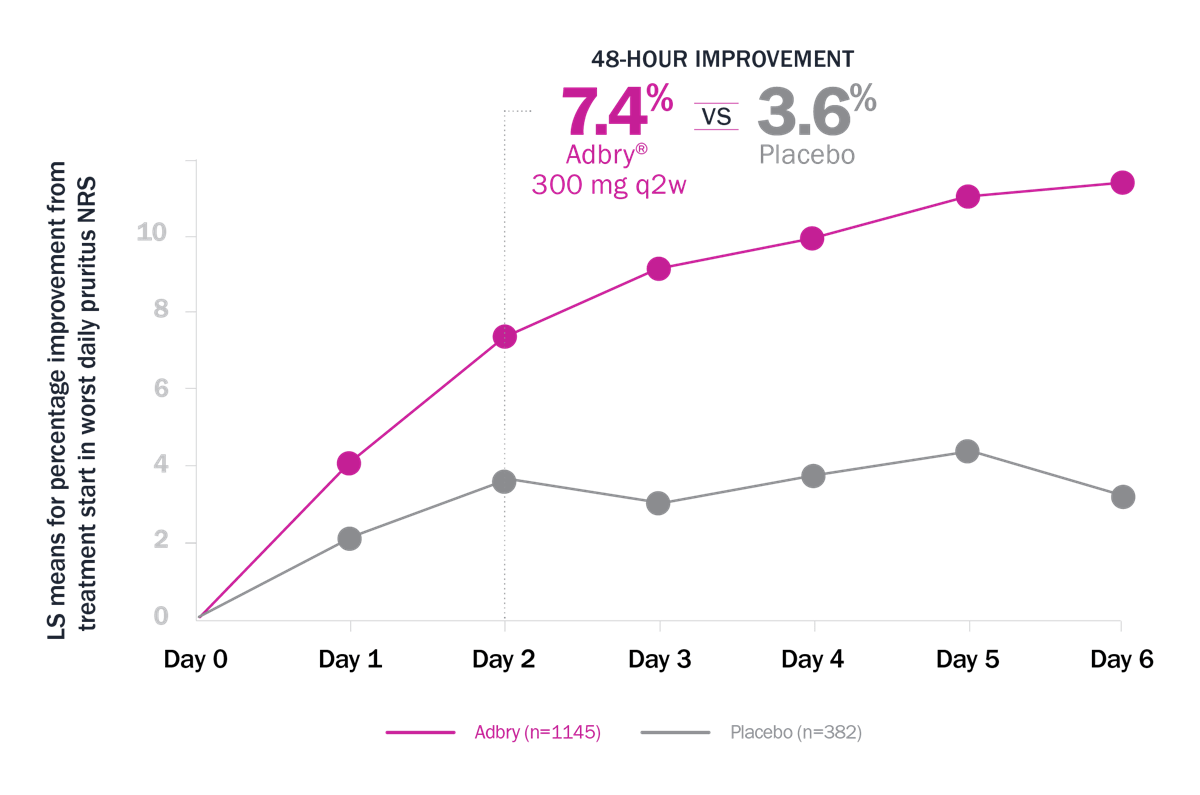
Limitations: Itch reduction as defined by ≥4-point reduction in worst daily pruritus NRS (weekly average) was a prespecified endpoint at Week 16 in each of the pooled studies. This analysis was not prespecified and not adjusted for multiplicity. Conclusions should be made with caution.
* LS means for % improvement from treatment start in Worst Daily Pruritus NRS: 7.4% with Adbry vs 3.6% with placebo at 48 hours.3 † Patients who received rescue treatment or with missing data were considered nonresponders.


Long-Term Results
Open-label extension results
The ECZTEND study demonstrated long-term results with up to 4 years of data4
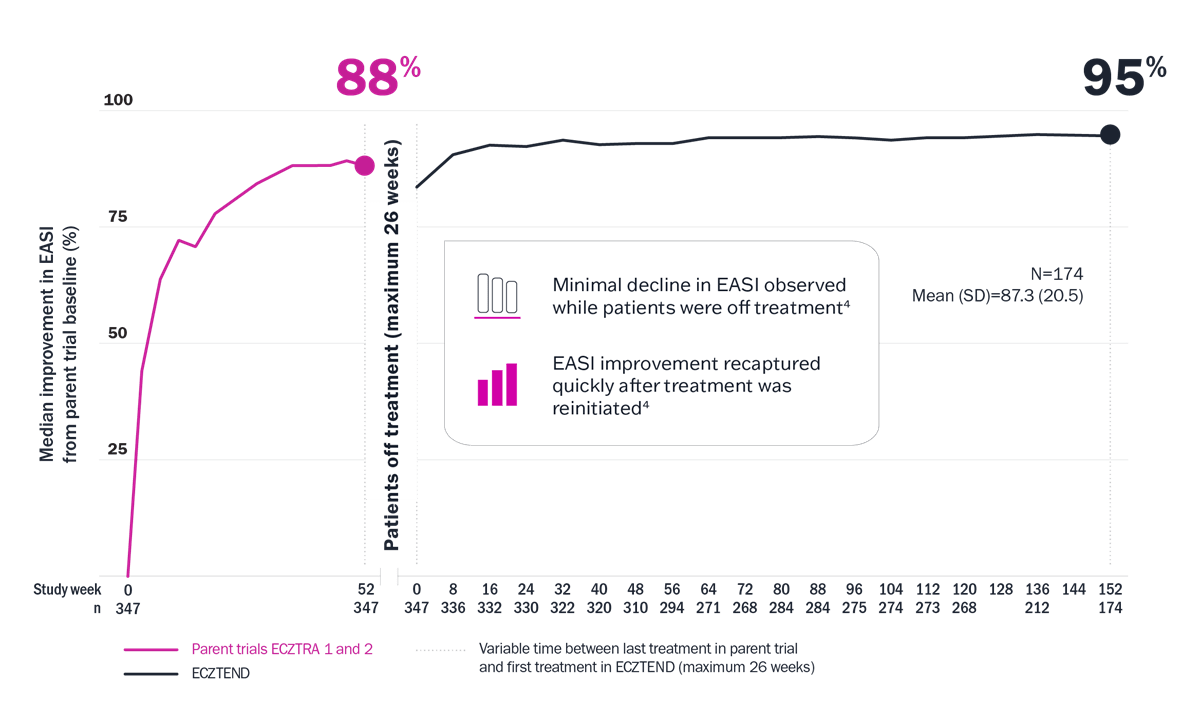
Limitations: Limitations and context associated with the open-label study design and data include decreasing sample size, potential continued involvement of responders, and attrition of nonresponders. Data presented are descriptive in nature and no statistical comparisons are made.
* Median EASI score at pivotal trial baseline: 26.7; at ECZTEND baseline: 4.7; at week 152: 1.4.3 † Variable time between last treatment in parent trial and first treatment in ECZTEND (maximum 26 weeks). ‡ A total of 2666 patients from the ECZTRA 1, 2, 3, 5, 6, 7, and 8 parent trials were enrolled in ECZTEND as of data cutoff. Data presented from a post-hoc interim analysis of an ongoing OLE trial represent a selected subgroup of patients (n=347) from the parent trials ECZTRA 1 and 2 who elected to enter the ECZTEND trial and had consistently received Adbry for a total of up to 4 years at data cutoff (April 30, 2022). As such, data may not be generalizable to the full Adbry population.
EASI=Eczema Area and Severity Index.
ECZTEND was a phase 3, long-term, 5-year, open-label, single-arm extension trial that evaluated the safety and efficacy of Adbry in patients with AD who participated in the previous Adbry parent trials. Patients were permitted to enter ECZTEND after completion of the parent trial regardless of their treatment response or whether they were treated with Adbry or placebo. Patients received a loading dose followed by 300 mg q2w plus optional TCS as needed.4
Visible Results
Results across body areas
Click the areas below to see data